|
|
|
|
|
R. Orlando |
| Introduction |
In this session we will discuss the way CRYSTAL [1]
can be used to study open-shell systems such as transition metal oxides in various
magnetic states (ferromagnetic (FM), antiferromagnetic (AFM), ferrimagnetic)
and how a system may be forced into the desired spin configuration.
| A simple magnetic compound: KMnF3 [2] |
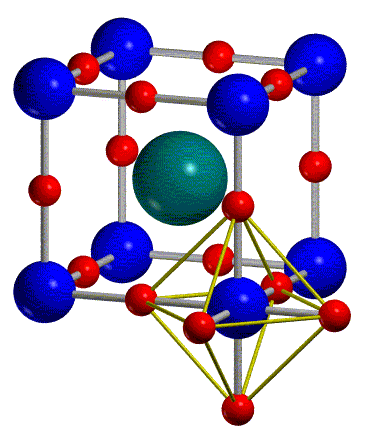
The geometry of KMnF3 (perovskite structure) is represented in figure 1: the space group is Pm-3m, there is one formula unit per cell and the asymmetric unit includes three atoms in the following special positions:
| Mn (0,0,0) | K (1/2.1/2,1/2) | F (1/2,0,0) |

|
From our point of view KMnF3 is appealing for several reasons:
Which are the observables we are interested in?
First of all, we want to compare the relative stability of the
FM and the AFM phases. On this purpose, we must run the code twice,
each time specifying the spin configuration in the input file.
The FM solution is obtained by requiring that there are 5 unpaired electrons per unit cell. Since the unit cell contains only one transition metal atom, this requirement implies that the total spin projection in the unit cell along the z direction (Szcell) is 5/2.
The CRYSTAL directive is SPINLOCK, which requires the specification of two integer arguments:
|
SPINLOCK
5 30 |
Keyword to lock the spin difference
na-nb
na-nb and number of SCF cycles with na-nb fixed |
i.e. populate states in such a way that
na-nb
= 5 (na and
nb denote the number of
Exercise 1: Run CRYSTAL with file kmnf3.d12 as an input.![]() -
-![]() spin electrons per cell)
for 30 SCF cycles and remove the constraint from the 31-st cycle on.
spin electrons per cell)
for 30 SCF cycles and remove the constraint from the 31-st cycle on.
Note: The SPINLOCK directive does not constrain unpaired electrons to localize at the Mn ions. It simply states that Szcell needs to be 5/2.
Answers:
Exercise 2: Verify that, when the SPINLOCK directive in exercise 1 is omitted, a diamagnetic phase is obtained with metallic properties.
Exercise 3: Repeat exercise 1 by restricting the SPINLOCK directive to one SCF cycle only.
Although, in principle, we could try an infinite number of AFM cells with
Szcell = 0
to find the most stable phase, fortunately, it is well known
that the experimental AFM phase of KMnF3 corresponds to that geometry
where each Mn ion is surrounded by six nearest neighbouring Mn ions with
opposite spin.
To achieve this, we must consider a cell containing two nearest
neighbouring Mn ions and break all symmetry relationship between them, so that
they may carry opposite spin (see figure 1), i.e. transform the primitive
cubic lattice into a commensurate face-centered cubic super lattice, which
results from interpenetrating ![]() and
and ![]() -spin
Mn sub lattices.
The easiest way to do it with CRYSTAL is to use the SUPERCEL(L) option to
define the geometry of the appropriate double volume cell.
The input cards to be added in the geometry input section, in this case,
are the following:
-spin
Mn sub lattices.
The easiest way to do it with CRYSTAL is to use the SUPERCEL(L) option to
define the geometry of the appropriate double volume cell.
The input cards to be added in the geometry input section, in this case,
are the following:
|
SUPERCEL
1 1 0 1 0 1 0 1 1 |
Keyword to create a super cell
Input of the expansion matrix elements |
The nine arguments to the SUPERCEL(L) option define the transformation
matrix by rows. The matrix is multiplied by column vectors from the left,
so that the new super lattice parameters (bi, = 1,2,3)
are expressed in terms of the primitive lattice parameters
(ai) as:
The bi vectors relate second nearest neighbouring Mn atoms to each
other (along the face diagonals), whereas next nearest neighbours
are no longer equivalent and can carry spin up and down, alternatively, as
is shown in the central picture of figure 1.
As the cell is doubled, the number of atoms per cell doubles too
(10 instead of 5). However, the symmetry remains cubic (48 point symmetry
operators).
In this case, the SPINLOCK directive will specify that
Szcell = 0:
Actually, this condition can be satisfied in two different ways: either
spin density is zero everywhere in the cell (diamagnetic or closed shell) or,
although spin density is
not zero, there are as many
Exercise 4: Run CRYSTAL for AFM KMnF3 specifying the
SUPERCEL(L) and SPINLOCK directives, as suggested above.
The reason for this nasty behaviour is that the initial guess Fock matrix
so defined is too far from the final SCF solution, in this case.
Therefore, it is always helpful - in many cases mandatory - to drive
the system to the required solution through the choice of a more reasonable
initial guess.
At least two alternative ways are available in CRYSTAL to serve this purpose:
a) start the SCF cycle from the isolated atom solution and use the
ATOMSPIN directive
b) start the SCF cycle from the corresponding
FM solution and use the SPINEDIT directive.
In this way CRYSTAL is asked to define open shell configurations for
two atoms in the initial guess:
the five unpaired electrons of the first atom in the output list (one
of the two Mn ions in the cell) are assigned spin
The SCF for the AFM case is started from the density matrix of the
corresponding FM phase, where spin inversion is applied to the unpaired
electrons of one of the Mn ions: the second atom in the output list.
Note: If the FM and AFM solutions are obtained with the same geometry,
the same set of mono- and bi-electronic integrals is used in both cases
(they need to be computed only once).
Exercise 5: Repeat exercise 4 adding the ATOMSPIN
directive in the SCF input section as specified above.
Exercise 6: Run CRYSTAL for the FM KMnF3
using the (double) super cell specified above, save fortran unit 9 and copy to unit 20,
then repeat exercise 4 adding the GUESSP and SPINEDIT directives
in the SCF input section as specified above.
Answer:
Exercise 7: Use the results of exercises 1 and
4 to calculate the FM-AFM energy difference for KMnF3.
Which is the more stable phase?
Is the method accurate enough for the calculation of
Note: One of the crucial points of computational solid state chemistry
and physics in this decade is the implementation of algorithms
able to provide high numerical accuracy.
If you have a periodic code at home, check one aspect
of its numerical accuracy by performing the following test:
evaluate the total energy of a system with a unit cell and with
a super cell. The total energy should
obviously scale linearly with the volume or the number of atoms.
The calculation of the magnetic exchange coupling constant J is
particularly important for comparison with experiment.
Ising's simple model of a spin lattice allows to relate J to
b1 = a1 + a2
b2 = a1 + a3
(1)
b3 = a2 + a3
SPINLOCK
0 30
Keyword to lock the spin difference
na-nb
na-nb
and number of SCF cycles with
na-nb
fixed
![]() - as
- as
![]() -spin electrons (any number).
With this indeterminacy it is usually difficult to converge the SCF cycle to
any definite state with CRYSTAL and, even if one succeeds in converging
to some solution, it will generally not be
any of the possible AFM phases, but most likely some metallic state with
much higher total energy per cell. This situation is worst for system with unpaired electron
in
-spin electrons (any number).
With this indeterminacy it is usually difficult to converge the SCF cycle to
any definite state with CRYSTAL and, even if one succeeds in converging
to some solution, it will generally not be
any of the possible AFM phases, but most likely some metallic state with
much higher total energy per cell. This situation is worst for system with unpaired electron
in
ATOMSPIN
2
1 +1 2 -1
Keyword to set the atomic spin
number of atoms to attribute a spin
atom labels and spin (1, 0, -1)
![]() (+1) and those
of the second atom in the list (the other Mn ion) spin
(+1) and those
of the second atom in the list (the other Mn ion) spin ![]() (-1).
If not specified, all equivalent atoms would be
attributed the same spin (
(-1).
If not specified, all equivalent atoms would be
attributed the same spin (![]() or
or
![]() ).
).
GUESSP
SPINEDIT
1
2
Keyword to read the density matrix from a previous run
Keyword to edit the spin density matrix
number of atoms which spin must be reversed
atom labels
Question:
The FM-AFM energy difference (
![]() E)
E)
![]() E?
E?
Since magnetic phase transitions involve quite small
![]() E, of the order of 10-4
hartree, special care must be paid in the
determination of the total energy for the two phases. In order to check
whether they are accurate enough to give meaningful energy differences,
the following tests can be done:
E, of the order of 10-4
hartree, special care must be paid in the
determination of the total energy for the two phases. In order to check
whether they are accurate enough to give meaningful energy differences,
the following tests can be done:
![]() E
determination?
E
determination?
We compare the total energy of the FM phase obtained in exercise 1
(unit cell, 1 Mn ion) with that obtained with the super cell defined by
vectors 1 (2 Mn ions per cell, 10 unpaired electrons):
-2047.643082 and -4095.286163 hartree, respectively. The super cell energy is
almost exactly twice the unit cell energy, the error is smaller than
1 .10-6 hartree, i.e. two orders of magnitude less than
![]() E.
E.
The small value of
![]() E certainly leads to exclude
a priori that any significant
geometry variation may take place during the FM-AFM transition,
so that both phases can be described as having essentially the same geometry.
On the other hand, as
E certainly leads to exclude
a priori that any significant
geometry variation may take place during the FM-AFM transition,
so that both phases can be described as having essentially the same geometry.
On the other hand, as
![]() E decreases with
the Mn-Mn distance very rapidly, small errors in the
determination of equilibrium geometry (typically of the order of
2% within the HF approximation) may affect
E decreases with
the Mn-Mn distance very rapidly, small errors in the
determination of equilibrium geometry (typically of the order of
2% within the HF approximation) may affect
![]() E
with large errors.
For these reasons we are discouraged from optimizing geometry, and
working at the experimental geometry is certainly to be preferred.
E
with large errors.
For these reasons we are discouraged from optimizing geometry, and
working at the experimental geometry is certainly to be preferred.
Data in table 1 show how
![]() E
varies when improving the basis set.
E
varies when improving the basis set.
![]() E)
of KMnF3 as a function of the basis set.
E)
of KMnF3 as a function of the basis set.
case
Basis set
DE
a
as used in exercise 1
0.293
b
case a + d on F
(
![]() =0.7)
=0.7)0.293
c
case a with 5-1d on Mn instead of 4-1d
0.292
d
case c + sp
(
![]() =0.25) on Mn
=0.25) on Mn0.334
e
case d + d
(
![]() =0.4) on K
=0.4) on K0.325
Case e can be considered close to the HF limit.
![]() E changes within 15% in the variational
basis set range considered. Basis set incompleteness may certainly
not be invoked as an explanation of the much larger discrepancies between the calculated and experimental
E changes within 15% in the variational
basis set range considered. Basis set incompleteness may certainly
not be invoked as an explanation of the much larger discrepancies between the calculated and experimental
![]() E.
E.
The magnetic exchange coupling constant
![]() E
[3,
4],
provided the unpaired electrons are fairly well localized and
the magnets interact as Ising magnets. In fact, Ising hamiltonian
[5]
is a particular case of Heisenberg spin hamiltonian where it is assumed
that spin-spin interaction takes place through only one magnetic component
(along the z direction):
E
[3,
4],
provided the unpaired electrons are fairly well localized and
the magnets interact as Ising magnets. In fact, Ising hamiltonian
[5]
is a particular case of Heisenberg spin hamiltonian where it is assumed
that spin-spin interaction takes place through only one magnetic component
(along the z direction):
| (2) |
where the spin operators ![]() are applied to ions at the R
and R' lattice nodes.
Although, in general, this is not the case, the application of the Ising model
is much easier than the use of Heisenberg hamiltonian and is more compatible
with our monodeterminantal solution, which corresponds to non pure spin state
for general open-shell systems. In addition, it has been shown
[6]
that eq. 2 is obtained from the Heisenberg hamiltonian
when the mean field approximation is applied.
are applied to ions at the R
and R' lattice nodes.
Although, in general, this is not the case, the application of the Ising model
is much easier than the use of Heisenberg hamiltonian and is more compatible
with our monodeterminantal solution, which corresponds to non pure spin state
for general open-shell systems. In addition, it has been shown
[6]
that eq. 2 is obtained from the Heisenberg hamiltonian
when the mean field approximation is applied.
How is eq. 2 further simplified?
|
|
(3) |
i.e.
![]() E is proportional to the number
(N) of symmetric Mn atoms in the (super)cell, the number
(Z) of the next nearest (Mn) neighbours to each of the N
Mn with opposite spin, the square of the Mn spin component
along the n direction. The proportionality constant is J.
E is proportional to the number
(N) of symmetric Mn atoms in the (super)cell, the number
(Z) of the next nearest (Mn) neighbours to each of the N
Mn with opposite spin, the square of the Mn spin component
along the n direction. The proportionality constant is J.
Exercise 8: Use the result of exercise 7 to calculate
J for the FM-AFM transition. ( Note: experimental J
are usually reported in Kelvin; the conversion factor to express
J in K from
![]() E
in atomic units is 3.1577.105).
E
in atomic units is 3.1577.105).
| Breaking symmetry: KCoF3 [2] |
Exercise 9: Run CRYSTAL with file kcof3.d12
as an input and discuss Mulliken population analysis data with particular
reference to the d atomic orbitals of Co.
The result of exercise 9 shows that spin density is large only at the d atomic orbitals of the transition metal, just as happens with KMnF3. Therefore, the simple model of a Co ion in a crystal field can be helpful in the interpretation of the transition metal electronic configuration in this case, too. According to crystal-field theory, the d-levels of each Co ion, at the centre of a regular octahedron formed by 6 F ions (point group: Oh, or m-3m), split into two sets of degenerate levels, which group theory classifies as t2g (triply degenerate) and eg (doubly degenerate), respectively. The dxy , dxz , dyz atomic orbitals are a basis for the t2g irreducible representation, d2z2-x2-y2 and dx2-y2 for eg and elementary electrostatics considerations suggest that t2g be lower in energy.
Is the result of exercise 9 correct?
Hund's rules say how the d levels of an atom must be populated: since the atomic
configuration of Co includes 7 electrons in the d orbitals, maximum spin
multiplicity is gained with two doubly- and three singly-occupied d levels.
Actually, Mulliken atomic orbital population data in CRYSTAL output
from exercise 9 do not comply with Hund's rules. In fact, due to
symmetry constraints only the two eg levels are singly-occupied
and degeneracy imposes that nearly 1.7 |e| are accommodated in each of the
t2g orbitals. A more stable
configuration can be obtained only by partly removing the t2g
degeneracy and allowing Jahn-Teller distortion. This means that crystalline
KCoF3 must have lower symmetry than cubic. For example,
if the CoF6 octahedron is distorted along one of its principal
directions, say z, symmetry is lowered to D4h
(4mmm) and the t2g
representation is no longer irreducible, so that it can be decomposed
into two terms: eg and b2g.
dxz and dyz are the basis for
the doubly degenerate eg and dxy
for the monodimensional representation.
This corresponds to the transformation from cubic symmetry to tetragonal.
How do we tell CRYSTAL to reduce symmetry?
One possibility is consulting the International Tables of Crystallography
to find the new space group in the list of the non-isomorphic subgroups of
Pm-3m. The group we are interested in is that
where all symmetry relationship between z
and the orthogonal coordinates x and y
are missing, i.e. P4mmm. This is the only piece of information
we need to prepare the new geometry input for CRYSTAL.
The other possibility is to cheat CRYSTAL. The ELASTIC
keyword in the geometry input section (see
CRYSTAL User's Manual)
allows distortion of the lattice
for the calculation of elastic constants. In this case, input of the following
cards:
|
ELASTIC
-2 0. 0. 0. 0. 0. 0. 0. 0. 0.000000001 |
Keyword to perform an elastic deformation of the lattice
deformation in terms of e strain tensor eij matrix elements |
has the effect of automatically reducing symmetry (to P4mmm, in this
particular example) without actually changing the lattice geometry.
In fact the matrix given in input is summed to identity and multiplied by
the starting lattice vectors from the left, so that, in this case, nor the
c lattice parameter is changed, being multiplied by 1.000000001.
Exercise 10: Repeat exercise 9 breaking lattice
symmetry (from cubic to tetragonal). Compare total energy and Mulliken
atomic orbital populations to those previously obtained.
Is there a way to converge CRYSTAL more rapidly to the
desired solution?
To speed up convergence in the SCF cycle the EIGSHIFT directive in the SCF
input section can be conveniently used as follows:
|
EIGSHIFT
1 1 6 5 0. .3 |
Keyword to alterate the orbital occupation before SCF
number of elements to be shifted (diagonal only) atom label, nr. of shells in the selected atom basis set, nr. of AO in the selected shell, a-spin shift, b-spin shift |
This directive allows to modify diagonal Fock matrix elements at the
beginning of the SCF cycle. If this is properly done, the system is
encouraged to converge to the solution corresponding to the hypothesized
electronic configuration. In this particular case, we identify the
first atom in the output list as Co, its sixth shell as that of the d atomic
orbitals and, among these, the fifth component of the d shell, i.e.
the dxy atomic orbital. We add 0.3 hartree to
the diagonal element corresponding to the dxy
atomic orbital in the ![]() -spin matrix and leave the
a-spin matrix unmodified.
-spin matrix and leave the
a-spin matrix unmodified.
Exercise 11: Repeat exercise 10 using the EIGSHIFT directive to speed up convergence.
Answers:
Exercise 12: Calculate
![]() E
(the FM to AFM transition energy) of KCoF3,
following the same procedure as for KMnF3.
E
(the FM to AFM transition energy) of KCoF3,
following the same procedure as for KMnF3.
The asymmetry introduced by Jahn-Teller distortion complicates the calculation of the magnetic coupling constants in this case. Since the nearest neighbouring Co ions of each Co along the z direction now differ from those on the xy-plane, two different coupling constants, Jxy and Jz, will correspondingly appear in eq. 2, when limiting the summation to six nearest neighbours. Eq. 3 can be applied to KCoF3 only if we consider an average J, otherwise a more appropriate equation is needed:
|
|
(4) |
where Zxy and Zz denote the number of the next nearest neighbours Co with opposite spin of a Co ion in and out of the xy-plane, respectively, i.e. Zxy = 4 and Zz = 2. The hypothesis of additivity suggests that the other relation that is necessary to calculate Jxy and Jz can be found by considering a different AFM phase. A good candidate is that phase with the spin configuration shown in the right picture of fig. 1, we refer to as AFM'. What shape has the super cell in this new lattice? A suitable choice is the following linear combination of the unit cell lattice vectors (ai):
| b1 = a1 | b2 = a2 | b3 = 2 a3 |
Exercise 13: Run CRYSTAL for AFM' KCoF3
and calculate the FM to AFM' energy difference
![]() E.
E.
From the application of eq. 2 to AFM' a new relation follows:
|
|
(5) |
Exercise 14: Calculate Jxy and Jz
from eqs. 4 and 5.
(by I. de P.R. Moreira)
Basis treateses on the general theory of magnetism: from introductory to more general aspects
of molecular and periodic magnetic systems:
Papers with applications of the CRYSTAL code to study several magnetic systems:
Thanks to R. Dovesi and I. de P.R. Moreira for reading the notes and for fruitful suggestions
|
|
|