|
|
|
|
|
B. Civalleri |
List of CRYSTAL geometry input examples
| Overview and goals |
Description of the geometry input section and the main optional keywords to edit the crystalline structure is presented. We primarily focus on systems with 3D periodicity: for 2D, 1D and 0D geometry refer to the CRYSTAL09 User's Manual. Note, however, that most of the 3D structures editing facilities can be applied to systems with lower periodicity.
Special attention will be given on how you can extract structural
information from crystallographic databases and literature papers.
Refer to CRYSTAL09 User's Manual
for complete description
of the geometry input section and related keywords.
For this tutorial we assume that you know
the basic concepts of crystallography.
See: P.Ugliengo, CRYSTALLOGRAPHIC STRUCTURAL
DATA.
This tutorial consists of two parts:
Example 1. A simple case: MgO, introduces the main features of the geometry input for a very simple inorganic system. Standard input and geometry editing keywords will be discussed.
Example 2. Molecular crystals: urea, discusses the case of a molecular crystal. CRYSTAL allows special editing of the geometry for molecular crystals.
A small collection of CRYSTAL geometry input examples is supplied.
Geometry input is read and processed by the program crystal.
The keyword TESTGEOM stops the run after reading the geometry input block and processing the data. It is inserted in all geometry input examples. It is useful to check crystal structures.
| Example 1. A simple case: MgO |
MgO crystallizes in a cubic cell with a
rock-salt structure. The crystal structure can be described as a fcc lattice
of Mg ions with O ions occupying all the octahedral holes or vice versa.
The rock-salt structure is the most common for MX compounds.
MgO is an important oxidic system in minerals,
in defective systems as well as in adsorption phenomena. Therefore, despite
its simplicity, MgO has been the subject of many research studies. Thus,
it represents a good example to show how geometry editing options
can be used with CRYSTAL09.
| COL
ICSD Collection Code 9863
DATE Recorded Jan 1, 1980; updated Jan 19, 1999 |
| NAME Magnesium oxide |
| MINR
Periclase
FORM Mg O = Mg O TITL X-ray determination of electron-density distributions in oxides, Mg O, Mn O, Co O, and Ni O, and atomic scattering factors of their constituent atoms REF Proceedings of the Japan Academy PJACA 55 (1979) 43-48 AUT Sasaki S, FujinoK, TakeuchiY SYM x, y, z y, z, x z, x, y x, z, y y, x, z z, y, x x, -y, -z y, -z, -x z, -x, -y x, -z, -y y, -x, -z z, -y, -x -x, y, -z -y, z, -x -z, x, -y -x, z, -y -y, x, -z -z, y, -x -x, -y, z -y, -z, x -z, -x, y -x, -z, y -y, -x, z -z, -y, x -x, -y, -z -y, -z, -x -z, -x, -y -x, -z, -y -y, -x, -z -z, -y, -x -x, y, z -y, z, x -z, x, y -x, z, y -y, x, z -z, y, x x, -y, z y, -z, x z, -x, y x, -z, y y, -x, z z, -y, x x, y, -z y, z, -x z, x, -y x, z, -y y, x, -z z, y, -x |
| CELL a=4.217(1) b=4.217(1) c=4.217(1) alpha=90.0 beta=90.0 gamma=90.0 |
| V=75.0 D=3.56 Z=4 |
| SGR F m -3 m (225) - cubic |
| CLAS
m-3m (Hermann-Mauguin) - Oh (Schoenflies)
PRS cF8 ANX AX PARM Atom__No OxStat Wyck -----X----- -----Y----- -----Z----- -SOF- |
|
Mg 1 2.000 4a 0.
0. 0.
O 1 -2.000 4b 0.5 0.5 0.5 |
| WYCK
b a
ITF Mg 1 B=0.312 ITF O 1 B=0.362 REM M PDF 43-1022 RVAL 0.013 |
The information you need to define the crystal structure is highlighted. Basically, the crystal structure is determined by the space group, by the shape and size of the unit cell and by the relative position of the atoms in the asymmetric unit.
MgO geometry input, derived from ICSD data, will be prepared and discussed, line by line.
1. Title section
MgO bulk: crystal structure from ICSD
The first line contains the title section. It can be useful to indicate the system in study and other relevant information about the job. The title section is printed in the output file, but it is not otherwise used by CRYSTAL.
2. Dimensionality of the system
CRYSTAL
The first record of the geometry definition
must specify the dimensionality of the system.
CRYSTAL adopts four keywords:
CRYSTAL, SLAB, POLYMER
and MOLECULE, for 3D, 2D, 1D and 0D systems, respectively.
In this case the keyword to specify is
CRYSTAL.
The keyword EXTERNAL allows geometry input from external file (see CRYSTAL User's Manual for further details).
3. Crystallographic information (for 3D systems only)
0 0 0
three integer numbers:
- convention
for the space group identification: sequential number (0) or alphanumeric
code (1).
- type of cell
for rhombohedral groups: hexagonal (0) or rhombohedral (1).
- setting of the origin (see CRYSTAL User's Manual for
further details).
4. Space group
225
It can be indicated either with its sequential number (0), as in this case, or by the Hermann-Mauguin alphanumeric code (1). In the ICSD file you can find both of them.
So, till now, the input file would look
something like this:
| MgO
bulk: crystal structure from ICSD
CRYSTAL 0 0 0 225 |
MgO
bulk: crystal structure from ICSD
CRYSTAL 1 0 0 F M 3 M |
according to the sequential number (on
the left) or the alphanumeric code (on the right).
Note: CRYSTAL adopts F M 3 M instead
of F M -3 M for compatibility with a previous edition of the International
Tables for Crystallography (see CRYSTAL User's Manual for further
details).
5. Lattice parameters
4.21
The minimal set of crystallographic cell parameters is indicated (in Angstrom and degrees). For MgO, cubic system, the length of the edge of the cell fully defines shape and size of the conventional unit cell (note, however, that CRYSTAL works on the primitive cell).
6. Atomic position specification
2
12
0.0 0.0 0.0
8
0.5 0.5 0.5
The first line gives the number of atoms in the asymmetric unit. One line per atom in the asymmetric unit follows, to specify the conventional atomic number and the coordinates in fractional units of the crystallographic lattice vectors. These atoms are usually indicated as non-equivalent atoms, i.e. atoms not symmetry related. The whole structure of MgO is defined by 2 atoms.
7. Closing the geometry input section
END
This keyword ends the geometry input section. Before ending the section, you may specify optional keywords to modify the structure.
The completed input file looks
like:
| MgO
bulk
CRYSTAL 0 0 0 225 4.21 2 12 0.0 0.0 0.0 8 0.5 0.5 0.5 |
1.
Title of the job
2. Dimensionality of the system 3. Crystallographic information (3D only) 4. Space Group number 5. Lattice parameters 6. Number of atoms in asymmetric unit Atomic position specification in fractionary coordinates |
|
| TESTGEOM | Optional keyword to stop execution after geometry input | |
| END | 7. end of the geometry input section |
Exercise:
Create a file mgo.d12 and type the
geometry input above. Visualize the structure following the instructions
reported in the How to run CRYSTAL section.
Exercise:
Here is reported an entry for a-alumina
from the ICSD database. Use the crystallographic information to prepare
the corresponding CRYSTAL geometry input file and visualize the structure.
| COL
ICSD Collection Code 73724
DATE Recorded Jan 10, 1995; updated Nov 10, 1997 NAME Aluminium oxide - alpha MINR Corundum FORM Al2 O3 = Al2 O3 TITL Synchrotron X-ray study of the electron density in alpha-Al2O3 REF Acta Crystallographica B (39,1983-) ASBSD 49 (1993) 973-980 AUT Maslen E N, Streltsov V A, Streltsova N R, Ishizawa N, Satow Y SYM x, y, z -y, x-y, z y-x, -x, z -y, -x, 1/2+z x, x-y, 1/2+z y-x, y, 1/2+z -x, -y, -z y, y-x, -z x-y, x, -z y, x, 1/2-z -x, y-x, 1/2-z x-y, -y, 1/2-z CELL a=4.754(1) b=4.754(1) c=12.982(1) alfa=90.0 beta=90.0 gamma=120.0 V=254.1 Z=6 SGR R -3 c H (167) - trigonal CLAS -3m (Hermann-Mauguin) - D3d (Schoenflies) PRS hR30 ANX A2X3 PARM Atom__No OxStat Wyck -----X----- -----Y----- -----Z----- -SOF- Al 1 3.000 12c 0. 0. 0.35223(4) O 1 -2.000 18e 0.69378(17) 0. 0.25 WYCK e c TF
Atom U(1,1) U(2,2) U(3,3) U(1,2) U(1,3)
U(2,3)
|
|
|
|
 |
 |
When running CRYSTAL with the previous
input for MgO you will get the following output.
******************************************************************************* * * * CRYSTAL06 * * Release : 1.0 * * cry06_060822 * * * * * * * * MAIN AUTHORS * * * * R. DOVESI(1), V.R. SAUNDERS(2), C. ROETTI(1), R. ORLANDO (1,3), * * C.M. ZICOVICH-WILSON(1,4), F. PASCALE(5), B. CIVALLERI(1), K. DOLL(6), * * N.M. HARRISON(2,7), I. J. BUSH(2), Ph. D'ARCO(8), M. LLUNELL(9) * * * * (1) THEORETICAL CHEMISTRY GROUP - UNIVERSITA' DI TORINO - TORINO (ITALY) * * http://www.crystal.unito.it * * (2) COMPUTATIONAL SCIENCE & ENGINEERING DEPARTMENT - CCLRC DARESBURY (UK) * * http://www.cse.clrc.ac.uk/cmg/CRYSTAL/ * * (3) UNIVERSITA' DEL PIEMONTE ORIENTALE - ALESSANDRIA (ITALY) * * (4) UNIVERSIDAD AUTONOMA DEL ESTADO DE MORELOS - CUERNAVACA (MEXICO) * * (5) UNIVERSITE' HENRI POINCARE' - NANCY (FRANCE) * * (6) TU BRAUNSCHWEIG - BRAUNSCHWEIG (GERMANY) * * (7) IMPERIAL COLLEGE - LONDON (UK) * * (8) UNIVERSITE' PIERRE ET MARIE CURIE - PARIS (FRANCE) * * (9) UNIVERSIDAD DE BARCELONA - BARCELONA (SPAIN) * ******************************************************************************* EEEEEEEEEE STARTING DATE 26 08 2006 TIME 22:28:47.0 TEST11 - MGO BULK |
The header of CRYSTAL reports
the CRYSTAL version and the main authors of the code.
The title section
from the input file follows.
|
CRYSTAL CALCULATION
(INPUT ACCORDING TO THE INTERNATIONAL TABLES FOR X-RAY CRYSTALLOGRAPHY) CRYSTAL FAMILY : CUBIC CRYSTAL CLASS (GROTH - 1921) : CUBIC HEXAKISOCTAHEDRAL SPACE GROUP (CENTROSYMMETRIC) : F M 3 M |
Summary of the crystallographic
information. The periodicity of the system is indicated.
|
LATTICE PARAMETERS (ANGSTROMS AND DEGREES) - CONVENTIONAL CELL
A B C ALPHA BETA GAMMA 4.21000 4.21000 4.21000 90.00000 90.00000 90.00000 NUMBER OF IRREDUCIBLE ATOMS IN THE CONVENTIONAL CELL: 2 INPUT COORDINATES ATOM AT. N.
COORDINATES
|
The lattice parameters of the conventional cell and the atomic position
of the atoms in the asymmetric unit, as given in input, are reported.
******************************************************************************* << INFORMATION >>: FROM NOW ON, ALL COORDINATES REFER TO THE PRIMITIVE CELL ******************************************************************************* |
LATTICE PARAMETERS (ANGSTROMS AND DEGREES) - PRIMITIVE CELL
A B C ALPHA BETA GAMMA VOLUME
2.97692 2.97692 2.97692 60.0000 60.0000 60.0000 18.65462
COORDINATES OF THE EQUIVALENT ATOMS (FRACTIONARY UNITS)
N. ATOM EQUIV AT. N. X Y Z
1 1 1 12 MG 0.00000000000E+00 0.00000000000E+00 0.00000000000E+00
2 2 1 8 O -5.00000000000E-01 -5.00000000000E-01 -5.00000000000E-01
NUMBER OF SYMMETRY OPERATORS : 48
|
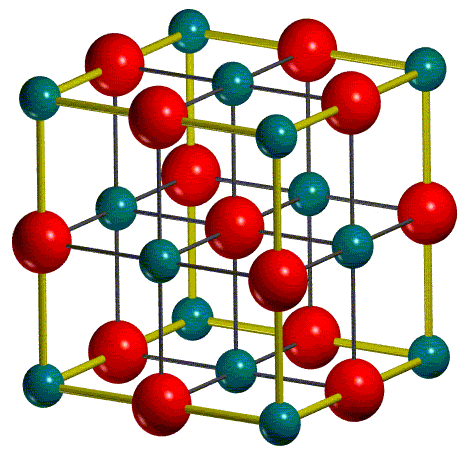
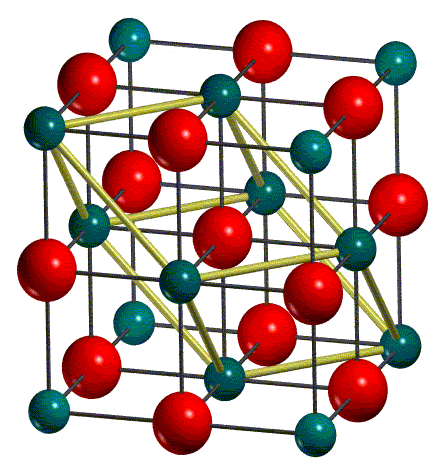
The crystallographic or conventional cell is used as standard option in input. It may be non-primitive, which means that it may not coincide with the cell of minimum volume (primitive cell) which contains just one lattice point. Note that, for maximum calculation efficiency, CRYSTAL works on the primitive cell. Hence, the conventional cell is transformed into the primitive cell (1/4 of the conventional cell), all the following structural information are referred to the primitive cell.
The transformation matrices conventional <=> primitive cell are given in Appendix A.4 of CRYSTAL03 User's Manual.
In the output file, the lattice parameters of the primitive cell and the corresponding atomic positions (in fractionary units) are reported. In this section, all the atoms in the primitive cell are displayed and indicated as equivalent atoms. For each non-equivalent atom the corresponding block of equivalent atoms is reported. For MgO two atoms only build up the primitive cell, as they are in special positions.
In figure, the conventional cell and the
primitive cell, enclosed in the conventional cell, of MgO are shown:
|
|
|
 |
 |
With the following banner ends the standard
CRYSTAL geometry output. After that, the output file continues with the
geometry output section relative to the optional keywords.
| *******************************************************************************
* GEOMETRY MANIPULATION - INPUT COORDINATES ARE GIVEN IN ANGSTROM ******************************************************************************* |
Exercise:
Compute the volume for the conventional
cell and compare it with the primitive cell. Which is the ratio between
the two cell volumes? Check the result on the printout.
Exercise:
Have a look at the output file for a-alumina
and examine the sections we have described above.
So far, we have considered the standard
CRYSTAL geometry input.
In the following section optional geometry keywords,
allowing structural editing, are presented.
Each keyword
will be presented by a specific example. So, for each example,
you can edit the input, run CRYSTAL and visualize the resulting structure.
The keyword SUPERCEL allows generation of a super cell by transformation of the lattice vectors of the primitive cell.
A super cell is obtained by defining the new unit cell vectors as linear combination of the primitive cell unit vectors. The new translation vectors b1', b2', b3' are defined in terms of the old vectors b1, b2, b3 and of the transformation matrix, E, read in input by rows, as follows:
b1'
= e11 b1 + e12b2
+ e13 b3
b2'
= e21 b1 + e22b2
+ e23 b3
b3'
= e31 b1 + e32b2
+ e33 b3
The symmetry is automatically reduced to
the point symmetry operators without translational components and a further
reduction of the symmetry is also possible. A shift of the origin to minimize
the number of symmetry operators with translational component is automatically
performed by crystal.
More information on the expansion matrix in "Defects"
Note: super cells can be generated for 2D
and 1D systems as well. In that case, the number of matrix elements decrease
from 9 to 4 and to 1, respectively.
For instance, the MgO primitive cell can
be transformed in the crystallographic one by the following matrix:
| -1.000
1.000 1.000
1.000 -1.000 1.000 1.000 1.000 -1.000 |
This corresponds to defining a quadruple cell.
Here is reported the corresponding CRYSTAL
geometry input:
| MGO
BULK super cell 8 atoms
CRYSTAL 0 0 0 225 4.21 2 12 0.0 0.0 0.0 8 0.5 0.5 0.5 |
Standard geometry input |
|
| SUPERCEL
-1.0 1.0 1.0 1.0 -1.0 1.0 1.0 1.0 -1.0 |
Keyword
for generating the
super cell
Input of the transformation matrix elements |
|
| TESTGEOM END |
Stop execution after geometry step End of the geometry input section |
Insert the highlighted lines in your MgO
input file and run CRYSTAL.
Looking at the output file you will find
the following section:
*******************************************************************************
* SUPERCELL OPTION
*******************************************************************************
EXPANSION MATRIX OF PRIMITIVE CELL
E1 -1.000 1.000 1.000
E2 1.000 -1.000 1.000
E3 1.000 1.000 -1.000
NUMBER OF ATOMS PER SUPERCELL 8
DIRECT LATTICE VECTORS COMPONENTS (ANGSTROM)
B1 4.210 0.000 0.000
B2 0.000 4.210 0.000
B3 0.000 0.000 4.210
LATTICE PARAMETERS (ANGSTROM AND DEGREES)
A B C ALPHA BETA GAMMA VOLUME
4.21000 4.21000 4.21000 90.0000 90.0000 90.0000 74.61846
**** ATOMS BELONGING TO THE SUPERCELL
LABEL AT.NO. COORDINATES (ANGSTROM AND FRACTIONARY)
1 12 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000
2 12 2.1050 2.1050 0.0000 0.5000 0.5000 0.0000
3 12 2.1050 0.0000 2.1050 0.5000 0.0000 0.5000
4 12 0.0000 2.1050 2.1050 0.0000 0.5000 0.5000
5 8 0.0000 0.0000 2.1050 0.0000 0.0000 0.5000
6 8 2.1050 2.1050 2.1050 0.5000 0.5000 0.5000
7 8 2.1050 0.0000 0.0000 0.5000 0.0000 0.0000
8 8 0.0000 2.1050 0.0000 0.0000 0.5000 0.0000
**************************** SUPERCELL GENERATED ****************************
|
The expansion matrix, given in input, the new lattice vectors components and lattice parameters, and the labels, atomic number, coordinates of the 8 atoms in the supercell are reported.
Atom label is the sequence number of the atom in the cell. In any geometry editing atoms are identified by their "label".
The new cell parameters correspond to the crystallographic lattice parameters. The new cell contains four MgO formula units.
Exercise:
Try to define larger super cells from the
MgO primitive cell, e.g. with 16 atoms, 32 atoms and so on. Hints in "Defects".
Exercise:
a-alumina has
a hexagonal unit cell but its primitive cell is rhombohedral, with volume
1/3 of the conventional cell. Starting from the rhombohedral cell, define
the expansion matrix to generate the crystallographic cell.
Hint for a better visualization:
The SUPERCEL keyword can be used
for visualizing larger fragments of the crystal structure when a molecular
visualizer is used.
The SUPERCEL option is a useful
starting point for interesting applications in materials science. For instance, such a keyword can be combined
with other options to define defective systems.
When a geometry editing (removal, insertion, substitution, displacement of atoms) modifies a site of the structure, the program can maintain or modify (reduce) the number of symmetry operators. Two keywords control the symmetry:
KEEPSYMM: In any subsequent editing of the geometry, the program will endeavour to maintain the number of symmetry operators. The symmetry operators are applied to the "perturbation", and if the multiplicity of the site is greater than 1, the perturbation will be multiplied by application of the symmetry operators.
BREAKSYMM [default]: subsequent modification of the geometry are allowed to alter (reduce: the number of symmetry operators can not be reduced) the point group symmetry. The new group is a subgroup of the original group and it is automatically obtained by crystal
The keyword SYMMREMO removes all point symmetry operators.
See keyword MODISYMM (input block 2, "Basis set", CRYSTAL03 User's Manual) for removal of selected symmetry operators.
The keyword ATOMSYMM prints the point group associated with each atomic position and the set of symmetry related atoms.
The keyword SYMMDIR prints the symmetry allowed directions, corresponding to the internal degrees of freedom (to obtain printing full crystal input must be submitted, block 1 2 3 4, with keyword TESTPDIM in block 3 - See CRYSTAL03 User's Manual)
MgO primitive cell - 48 symmetry operators:
ATOM 1 ATOMIC NUMBER 12 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 48 NO EQUIVALENT ATOMS ATOM 2 ATOMIC NUMBER 8 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 48 NO EQUIVALENT ATOMS THERE ARE NO SYMMETRY ALLOWED DIRECTIONS |
*******************************************************************************
ATOMS IN THE ASYMMETRIC UNIT 5 - ATOMS IN THE UNIT CELL: 16
ATOM X/A Y/B Z/C
*******************************************************************************
1 T 12 MG 0.000000000000E+00 0.000000000000E+00 0.000000000000E+00
2 T 12 MG -1.704907916233E-17 1.704907916233E-17 -5.000000000000E-01
3 F 12 MG -1.704907916233E-17 -5.000000000000E-01 1.704907916233E-17
4 F 12 MG 7.692414413785E-17 -5.000000000000E-01 -5.000000000000E-01
5 F 12 MG -5.000000000000E-01 -1.704907916233E-17 1.704907916233E-17
6 F 12 MG -5.000000000000E-01 7.272286071947E-17 -5.000000000000E-01
7 F 12 MG -5.000000000000E-01 -5.000000000000E-01 -1.085557425201E-17
8 T 12 MG 5.000000000000E-01 -5.000000000000E-01 -5.000000000000E-01
9 T 8 O -2.500000000000E-01 -2.500000000000E-01 2.500000000000E-01
10 T 8 O -2.500000000000E-01 -2.500000000000E-01 -2.500000000000E-01
11 F 8 O -2.500000000000E-01 2.500000000000E-01 2.500000000000E-01
12 F 8 O -2.500000000000E-01 2.500000000000E-01 -2.500000000000E-01
13 F 8 O 2.500000000000E-01 -2.500000000000E-01 2.500000000000E-01
14 F 8 O 2.500000000000E-01 -2.500000000000E-01 -2.500000000000E-01
15 F 8 O 2.500000000000E-01 2.500000000000E-01 2.500000000000E-01
16 F 8 O 2.500000000000E-01 2.500000000000E-01 -2.500000000000E-01
T = ATOM BELONGING TO THE ASYMMETRIC UNIT
ATOM 1 ATOMIC NUMBER 12 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 48 NO EQUIVALENT ATOMS ATOM 2 ATOMIC NUMBER 12 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 8 NUMBER OF ATOMS EQUIVALENT BY SYMMETRY 5 SEQUENCE NUMBERS OF THESE ATOMS 7 5 3 4 6 ATOM 3 ATOMIC NUMBER 12 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 8 NUMBER OF ATOMS EQUIVALENT BY SYMMETRY 5 SEQUENCE NUMBERS OF THESE ATOMS 6 2 5 4 7 . . . . . . . . . . . . . . ATOM 8 ATOMIC NUMBER 12 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 48 NO EQUIVALENT ATOMS ATOM 9 ATOMIC NUMBER 8 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 8 NUMBER OF ATOMS EQUIVALENT BY SYMMETRY 5 SEQUENCE NUMBERS OF THESE ATOMS 16 14 12 11 13 ATOM 10 ATOMIC NUMBER 8 - NUMBER OF SYMMOPS WHICH DO NOT MOVE THE ATOM 24 NUMBER OF ATOMS EQUIVALENT BY SYMMETRY 1 SEQUENCE NUMBERS OF THESE ATOMS 15 . . . . . . . . . . . . . SYMMETRY ALLOWED INTERNAL DEGREE(S) OF FREEDOM: 1 SYMMETRY ALLOWED DIRECTION 1 ATOM 9 (Z= 8) 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.4082483 0.0000000 0.0000000 0.0000000 -0.4082483 0.0000000 0.0000000 0.0000000 0.4082483 0.0000000 0.0000000 -0.4082483 0.0000000 0.4082483 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 0.0000000 -0.4082483 |
NEIGHBORS OF THE NON-EQUIVALENT ATOMS
N = NUMBER OF NEIGHBORS AT DISTANCE R
ATOM N R/ANG R/AU NEIGHBORS (ATOM LABELS AND CELL INDICES)
1 MG 6 2.1050 3.9779 9 O 0 0 0 11 O 0 0 0 12 O 0 0 0
13 O 0 0 0 14 O 0 0 0 16 O 0 0 0
|
The unique degree of freedom corresponds to the displacement of the first neighbours Oxygens.
After geometry editing, the symmetry is recognized through the symmetry operators, but the space group is not known. The program findsym (Harold T. Stokes and Dorian M. Hatch, package ISOTROPY ) identifies the space group of a crystal 3D, given the positions of the atoms in a unit cell. Input data are written by keyword FINDSYM in file FINDSYM.DAT, and saved as inpfilename.FINDSYM by the script runcry09.
Substitution of atoms
A simple example of a defective systems are substitutional defects. For instance, CRYSTAL has been used to study the energy of formation of a Ca defect in MgO: C. Freyria-Fava, R. Dovesi, V.R. Saunders, M. Leslie and C. Roetti, ``Ca and Be substitution in bulk MgO: ab initio Hartree-Fock and ionic model supercell calculation'', J. Phys.: Cond. Matter 5, 4793-4804 (1993).
For this purpose a 16 atoms super cell
of MgO was adopted. Here is reported the corresponding geometry
input section:
| SUPERCEL
2. 0. 0. 0. 2. 0. 0. 0. 2. |
Keyword
for generating the
super cell
Input of the expansion matrix elements |
|
| ATOMSYMM ATOMSUBS 1 1 20 |
Keyword
for analyzing the site symmetry Keyword for substituting atoms Number of atoms to be substituted Label of the atom to substitute, atomic number of the new atom |
|
| END | End of the geometry input section |
The keyword ATOMSUBS allows substitution of selected atoms in the cell, as defined when the keyword is entered. The total number of atoms to be substituted and, for each atom, the corresponding label and the new atomic number must be indicated.
Insert the lines shown above in your MgO
input file and run CRYSTAL.
******************************************************************************* * SUBSTITUTION OF 1 ATOM(S) ******************************************************************************* ATOM N. 1 MG (Z= 12) SUBSTITUTED WITH CA (Z= 20) |
In the output file the section above is displayed. Note: ATOM N. stands for atom number.
So, the procedure to define a substitutional defect is:
Exercise:
Starting from the silicon
CRYSTAL geometry
input, generate a super cell and create a carbon substitutional defect.
(Si data: cubic system, space group 227, a=5.42; one atom in the asymmetric unit
in 1/8, 1/8, 1/8, standard origin setting).
Another example of defective systems are vacancies. One of them is the so-called trapped-electron-hole centre. In such centres there are very often charge compensating impurities, e.g. a H atom. In MgO, this kind of defects are denoted as MgO:[H]0. The H atom formally substitutes a Mg atom at its lattice position and migrates towards one of the neighbouring O atoms, forming a strong covalent bond with it. The hole localizes at the opposite O atom completely. This defective system has been the subject of a recent paper.
In order to create the final defective
structure the initial MgO primitive cell have to be modified with a series
of geometry keywords. The new part of the input file looks something like:
| SUPERCELL
3. -1. -1. -1. 3. -1. -1. -1. 3. |
Keyword
for generating the
super cell
Input of the expansion matrix elements |
|
| ATOMSUBS
1 1 1 |
Keyword
for substituting atoms
Number of atoms to be substituted Label of the atom to substitute, atomic number of the new atom |
|
| ATOMDISP
3 1 0. 0. 1.131 19 0. 0. -0.005 17 0. 0. -0.100 |
Keyword
for displacing atoms - symmetry reduction allowed (default)
Number of atoms to be displaced Label of the atoms to displace, displacements in cartesian coordinates (Angstrom) |
|
| KEEPSYMM | Keyword for maintaining the symmetry in the following manipulations | |
| ATOMDISP
1 21 0.11 0. 0.004 |
Atom 21 |
|
| TESTGEOM END |
End of the geometry input section |
Insert the new lines in the MgO bulk input
and run CRYSTAL.
******************************************************************************* * DISPLACEMENT OF 3 ATOMS ******************************************************************************* ATOM N. 1 AT. N. 1 DISPLACED BY (A) 0.00000 0.00000 1.13100 ATOM N. 19 AT. N. 8 DISPLACED BY (A) 0.00000 0.00000 -0.00500 ATOM N. 17 AT. N. 8 DISPLACED BY (A) 0.00000 0.00000 -0.10000 THE NUMBER OF SYMMETRY OPERATORS HAS BEEN REDUCED FROM 48 TO 8 ******************************************************************************* |
Note that, in this example, the first atomic
displacement has been done allowing a symmetry reduction. Indeed, the number
of symmetry operators changes from 16 to 8 (see above). Whereas,
after KEEPSYMM, in the second displacement, the current symmetry is maintained
and all the atoms related by symmetry are moved (see below).
******************************************************************************* * DISPLACEMENT OF 1 ATOMS ******************************************************************************* ATOM N. 21 AT. N. 8 DISPLACED BY (A) 0.11000 0.00000 0.00400 SYMMETRY MAINTAINED - 21 ATOM(S) SYMMETRY-RELATED : SEQUENCE NUMBER OF ATOM(S) 31 24 32 OLD COORDINATES (FRAC. UNITS) 0.00000 0.25000 0.25000 NEW COORDINATES (FRAC. UNITS) 0.00048 0.26354 0.26306 |
A 32 atoms super cell has been used as starting
perfect cell and the defective center has been created substituting a Mg
atom by a H atom. Atoms 17 and 19, Oxygens, are 2 of the six equivalent first
neighbors of the atom at the origin. When H is at the origin, H
moves towards Oxygen 19, to form an OH group.
ATOMDISP allows displacement of selected
atoms in the cell as defined when the keyword is entered. The point symmetry
of the system is reduced.
In summary, for this example, a 32 atoms super cell of MgO has been adopted. The Mg atom at the origin is substituted with a hydrogen atom. Some atomic positions are changed in two steps. First, the position of the three atoms involved in the defect center (O-H...O) are displaced, allowing a reduction of the symmetry, then, preserving the symmetry (keyword KEEPSYMM), the neighbor oxygen atoms are relaxed. ATOMDISP is used to relax the atoms of the defect zone, keeping the maximum symmetry compatible with the model of the defect.
Exercise:
Instead of a hydrogen atom, the trapped-hole
may contain other cations. Define a defect structure with Li instead of
H.
Another useful keyword related to ATOMINSE
is ATOMREMO, which removes selected atoms from the primitive cell.
The keyword SLABCUT allows generation of a slab of given thickness, parallel to a plane of the 3D lattice specified by the h ,k , l crystallographic Miller indices.
Not all crystalline surfaces are physically stable or worthy of investigation. This is specially true for ionic or semi-ionic crystals. As an example, you may create a polar surface. It consists of charged layers alternating in such a way that the repeat unit has a net dipole per unit area, normal to the surface. Such surfaces are unstable and can only be prepared with substantial reconstruction or with the adsorption of charged species. See, for instance, the MgO (111) surface.
A section is dedicated to "Surfaces and adsorption". This section describes how to simulate a crystalline surface with CRYSTAL.
CRYSTAL
test cases 04-24, 05-25, 06-26, 07-27 show how to obtain the same 2D
structure with 2D input (04,05,06,07) and 3D input + cutting of a slab (24, 25,
26, 27)
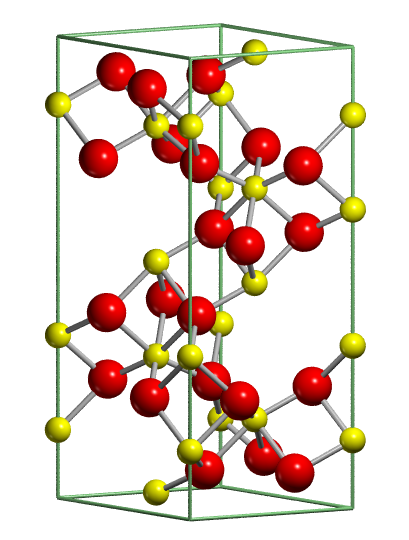
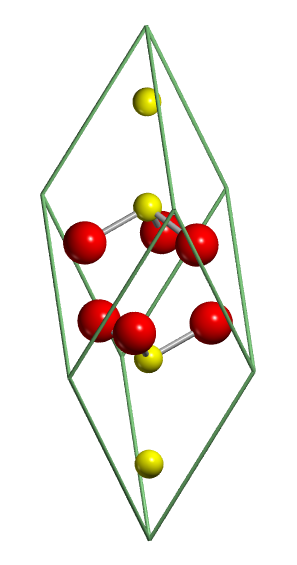
| Example 2. Molecular crystals: urea |
For molecular crystals special optional geometry keywords are available. As an example the urea crystalline structure will be used to illustrate such keywords.
The crystal structure of urea has a tetragonal
cell with four molecules in the unit cell (see figure below, on the left).
The molecules are linked to each other through hydrogen bonds, so as to
form infinite planar tapes. The arrangement of tapes is depicted in the
figure below on the right. They are mutually orthogonal, the cohesion among
them is provided by hydrogen bonds. Hence, each oxygen is involved in four
nearly equivalent hydrogen bonds, two within the tape and two with neighbouring tapes. Notice that the molecules within adjoining tapes are oppositely
oriented with respect to the crystallographic c axis; this provides
a further source of tape binding through dipole-dipole forces.
|
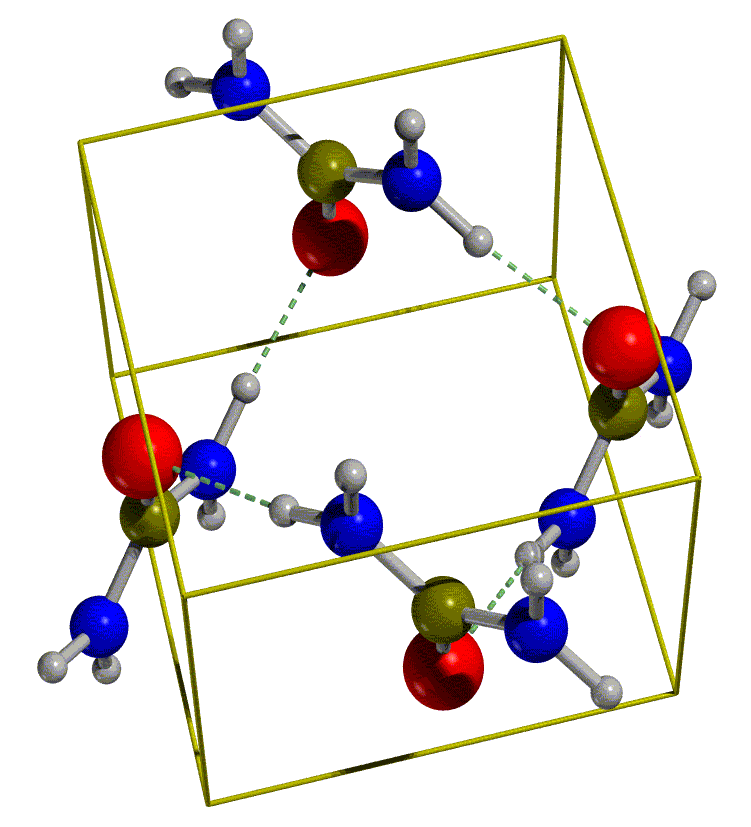 |
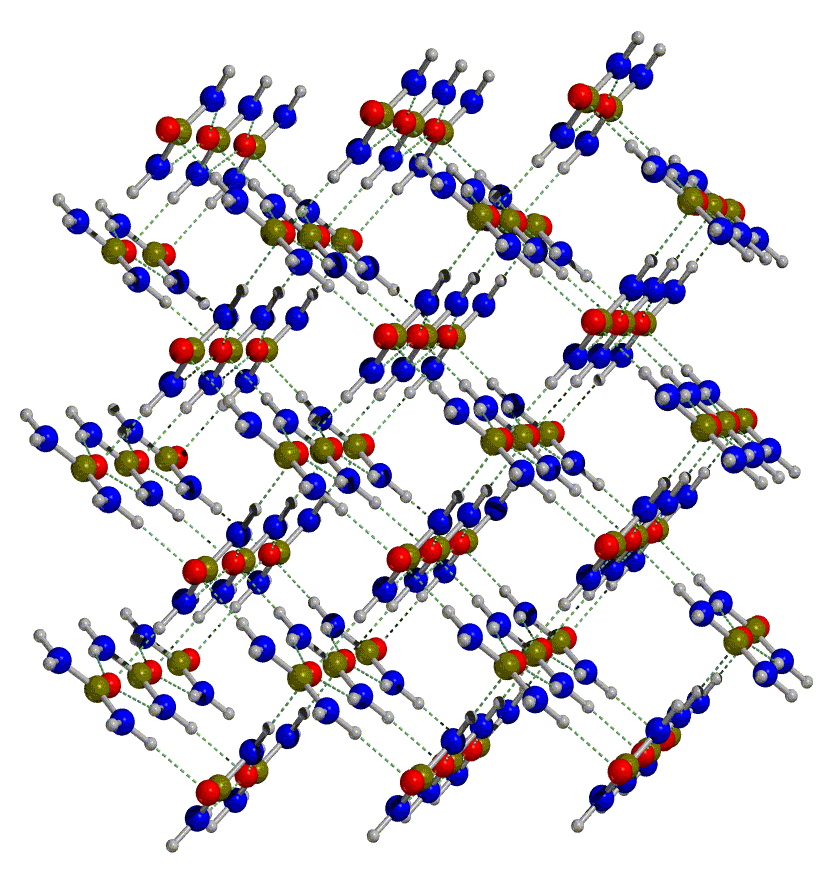 |
As for MgO, let us start from crystallographic data. Structural data on carbon-containing crystals can be find either on specific databases such as the Cambridge Structure Database (CSD) or directly from research articles and books. In the following, urea crystallographic data are reported from both of resources.
CSD entry:
| #UREAXX1243850207
10 9 0 0 0 8 4 5 3
0 8132200000110000000000084
5565 5565 4684 90 90 90333000 1 1 1 0 0 0 0 0113P-421m 240 R=0.0250 211 0121 0112 0101 0211 0110 0011 0101 0112 0121 0011 0110 0011 6121 6110 0 121 6211 6112 0211 6101 6110 0101 6011 6112 0 C 68H 23N 68O 68 C1 0 50000 32600 O1 0 50000 59530 N1 14590 64590 17660 H1 25750 75750 28270 H2 14410 64410 -3800 N1B -14590 35410 17660 H1B -25750 24250 28270 H2B -14410 35590 -3800 2 0 1 3 3 1 6 6 |
Crystal data from a resource book of crystal structure:
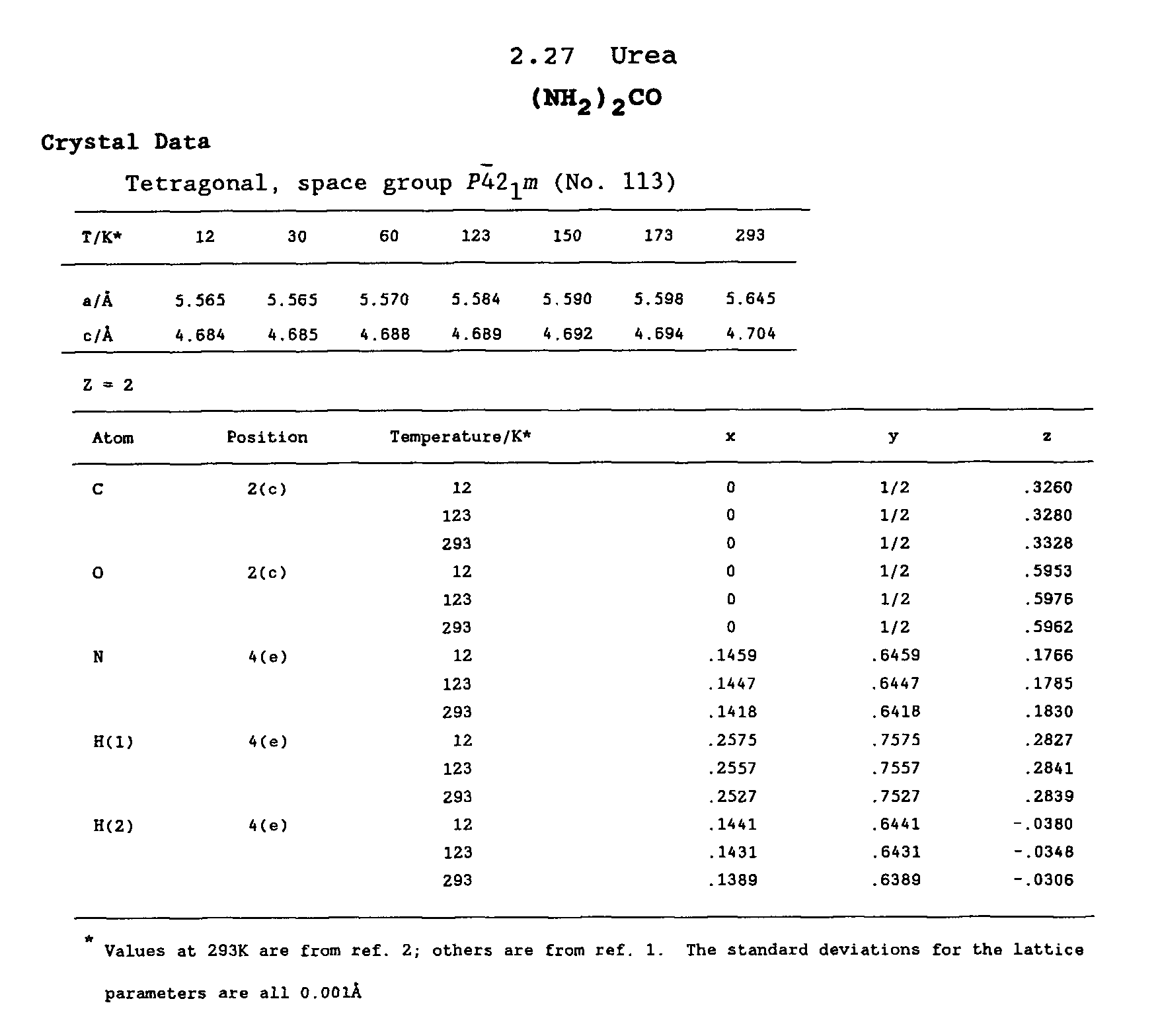 |
CSD crystal data corresponds to the urea
structure determined at 12 K. As noticed in the discussion on the reliability
of crystallographic structural data, structures which have been solved
at low temperature are to be preferred over those determined at room temperature.
In the table above you can assess the
dependence of the crystal data upon the temperature.
Hence, by using the structural data obtained
at 12K the CRYSTAL geometry input for urea can be prepared:
| UREA BULK
CRYSTAL 0 0 0 113 5.565 4.684 5 6 0.0000 0.5000 0.3260 8 0.0000 0.5000 0.5953 7 0.1459 0.6459 0.1766 1 0.2575 0.7575 0.2827 1 0.1441 0.6441 -0.0380 END |
All of the previously reported keywords may be used to modify the crystal structure of molecular crystals (e.g. SUPERCEL, ATOMDISP, ATOMINSE, SLABCUT, ...).
Exercise:
Try to use the SUPERCEL keyword
to visualize a large portion of the urea crystal structure.
However, few other keywords are specific
for molecular crystals.
This is the case of the MOLEXP
keyword. It allows to modify the cell parameters according to increments
given in input. However, although the volume of the cell is modified, the
symmetry and the internal geometry of the molecules in the cell are kept.
The urea input with MOLEXP is reported
below:
| UREA
BULK
CRYSTAL 0 0 0 113 5.565 4.684 5 6 0.0000 0.5000 0.3260 8 0.0000 0.5000 0.5953 7 0.1459 0.6459 0.1766 1 0.2575 0.7575 0.2827 1 0.1441 0.6441 -0.0380 |
Standard geometry input for urea bulk |
|
| MOLEXP
0.5 0.5 |
Keyword
to change the lattice parameters at constant symmetry and molecular geometry
Increments of the minimal set fo crystallographic parameters (e.g. tetragonal) |
|
| END |
Here the lattice parameters of the urea primitive cell
are expanded by 0.5 Angstrom. The corresponding
output is shown below:
| OLD LATTICE
- DIRECT LATTICE VECTOR COMPONENTS (BOHR)
5.56500 .00000 .00000 .00000 5.56500 .00000 .00000 .00000 4.68400 LATTICE
PARAMETERS (BOHR AND DEGREES) - PRIMITIVE CELL
LATTICE
PARAMETERS VARIATION (ANGSTROMS AND DEGREES) - CONVENTIONAL CELL
LATTICE
PARAMETERS (BOHR AND DEGREES) - PRIMITIVE CELL
|
After a classification of the molecular fragments in the primitive cell, the old lattice vectors are changed according to the increment given in input, and the new cell parameters are shown.
Exercise:
Insert the MOLEXP input section
in your urea geometry input, run CRYSTAL and visualize the modified crystal
structure. Notice how the internal molecular geometry is preserved by changing
the volume.
For molecular crystals an important geometry
manipulation is the extraction of molecular fragments from the bulk structure.
In CRYSTAL this task is accomplished by the MOLECULE keyword.
This option allows to extract one (or more) molecules from a molecular
crystal on the basis of chemical connectivity, defined by the sum of covalent
radii.
Note: For a proper identification of the
molecules it may be necessary a modification of the atomic radii (e.g.
when there are very short H-bonds linking the molecules in the lattice).
This can be done by using the RAYCOV option (see the CRYSTAL
User's Manual for further details)
In order to use the MOLECULE option two separate runs are necessary:
Step 1. Identification of molecular
fragments in the cell
The first step consists in a search of
the molecular fragments in the primitive cell, based on the chemical connectivity.
Accordingly, the atoms are reordered on the basis of the new classification.
In order to do that, the ATOMORDE keyword must be used. No input
data are required.
The urea input file looks something like:
| UREA
BULK
CRYSTAL 0 0 0 113 5.565 4.684 5 6 0.0000 0.5000 0.3260 8 0.0000 0.5000 0.5953 7 0.1459 0.6459 0.1766 1 0.2575 0.7575 0.2827 1 0.1441 0.6441 -0.0380 |
Standard geometry input for urea bulk |
|
| ATOMORDE | Keyword to classify molecular fragments in the cell | |
| END |
Here the ATOMORDE output section
is reported:
| *******************************************************************************
* SEARCHING THE MOLECULES OF THE CRYSTAL ******************************************************************************* MOLECULAR FRAGMENT
N. 1
MOLECULAR FRAGMENT
N. 2
******************************************************************************* |
Each molecular fragment is identified and the atoms are reordered fragment by fragment. Notice that further manipulations will refer to the new atomic numbering.
Step 2. Definition of the molecular
fragment
The second step concerns the definition
of the molecular fragment by means of the MOLECULE keyword. Now
we know the necessary information to run MOLECULE. In input, the
number of molecules to be extracted must be specified as well as a label
of an atom for each molecule and the integer coordinates of the cell where
that atom is positioned.
As an example, the geometry input for
the definition of an isolated urea molecule and the corresponding output
section are reported:
|
UREA BULK
CRYSTAL 0 0 0 113 5.565 4.684 5 6 0.0000 0.5000 0.3260 8 0.0000 0.5000 0.5953 7 0.1459 0.6459 0.1766 1 0.2575 0.7575 0.2827 1 0.1441 0.6441 -0.0380 ATOMORDE |
Standard geometry input for urea bulk |
|
| MOLECULE
1 1 0 0 0 |
Keyword
for extracting molecules from molecular crystals
Number of molecules to be extracted Label one atom in the molecule, integer coordinates of the cell containing the starting atom |
|
| END |
| *******************************************************************************
* MOLECULAR CALCULATION ******************************************************************************* MOLECULAR FRAGMENT
N. 1
******************************************************************************* |
On the basis of the previous re-ordering,
the molecular structure is defined and then a molecular calculation begins.
Edit the input reported above, run CRYSTAL
and visualize the isolated molecule.
When dealing with more than one molecule,
it may be difficult to identify the proper molecular fragments to be extracted.
A useful tool is to print a full neighboring analysis of the non-equivalent
atoms. By default, every full run, CRYSTAL06 prints up to 6 stars of neighbours.
The default value may be changed by means of the NEIGHBOR keyword.
Note: In order to use NEIGHBOR
you need a complete CRYSTAL input.
As an example, for urea bulk, the following
section is printed in the output file:
NEIGHBORS OF THE NON-EQUIVALENT ATOMS
N = NUMBER OF NEIGHBORS AT DISTANCE R
ATOM N R/ANG R/AU NEIGHBORS (ATOM LABELS AND CELL INDICES)
1 C 1 1.2614 2.3837 2 O 0 0 0
1 C 2 1.3447 2.5411 3 N 0 1 0 4 N 0 1 0
1 C 2 2.0367 3.8488 6 H 0 1 0 8 H 0 1 0
1 C 2 2.0477 3.8696 5 H 0 1 0 7 H 0 1 0
1 C 2 2.6461 5.0004 14 H 0 1 1 16 H 1 0 1
1 C 2 3.1089 5.8750 13 H 0 1 0 15 H 1 0 0
2 O 1 1.2614 2.3837 1 C 0 0 0
2 O 2 1.9922 3.7647 14 H 0 1 1 16 H 1 0 1
2 O 2 2.0582 3.8895 5 H 0 1 1 7 H 0 1 1
2 O 2 2.2726 4.2946 3 N 0 1 0 4 N 0 1 0
2 O 2 2.5002 4.7246 6 H 0 1 0 8 H 0 1 0
2 O 2 2.9550 5.5842 3 N 0 1 1 4 N 0 1 1
3 N 1 1.0053 1.8997 5 H 0 0 0
3 N 1 1.0092 1.9070 6 H 0 0 0
3 N 1 1.3447 2.5411 1 C 0-1 0
3 N 1 2.2726 4.2946 2 O 0-1 0
3 N 1 2.2965 4.3397 4 N 0 0 0
3 N 1 2.4939 4.7127 7 H 0 0 0
5 H 1 1.0053 1.8997 3 N 0 0 0
5 H 1 1.7473 3.3019 6 H 0 0 0
5 H 1 2.0477 3.8696 1 C 0-1 0
5 H 1 2.0582 3.8895 2 O 0-1-1
5 H 1 2.2682 4.2862 7 H 0 0 0
5 H 1 2.4939 4.7127 4 N 0 0 0
6 H 1 1.0092 1.9070 3 N 0 0 0
6 H 1 1.7473 3.3019 5 H 0 0 0
6 H 1 1.9922 3.7647 10 O 0 0 1
6 H 1 2.0367 3.8488 1 C 0-1 0
6 H 2 2.4984 4.7214 13 H 1 0 0 15 H 1 0 0
6 H 1 2.5002 4.7246 2 O 0-1 0
|
For each non-equivalent atom information on the first six (default) neighbours is printed: number, type, distance, position (indices of the cell). For instance C1 is linked to O2 in the same cell at 1.2614 Angstrom. Whereas, O2 is linked to H14, which is in the next cell, by an H-bond.
Exercise:
In the urea crystal structure, each oxygen
is involved in four nearly equivalent hydrogen bonds. Use the previous neighbouring
analysis to isolate a molecular fragment that contains the
four H-bonds.
When dealing with molecular crystals an important observable is the interaction energy. For a H-bonded molecular crystal as urea, the interaction energy is usually referred to the number of H-bonds. Basically, it coincides with the formation energy of the crystal per hydrogen bond with respect to the molecule in gas-phase.
Two optional keywords may help in computing
the interaction energy:
MOLSPLIT and MOLEBSSE. Even if they
are involved in the total energy calculation they must be specified in
the geometry input section.
The keyword MOLSPLIT performs an
expansion of the lattice, in such a way that the molecules of the crystal
are at an infinite distance from each other. No input data are required.
MOLEBSSE computes the BSSE in molecular
crystals via the counterpoise method. A molecular calculation is performed
with a basis set including the basis functions of the selected molecules
and the neighbouring atoms. The corresponding input section is reported
below:
| MOLEBSSE
1 1 0 0 0 3 4. |
Keyword
for the BSSE estimate in molecular crystals
Label one atom in the molecule, integer coordinates of the cell containing the starting atom Maximum number of stars of neighbours, maximum distance explored |
Note: Before running MOLEBSSE use
ATOMORDE
to identify the molecular fragments in the primitive cell.
Here is a summary of the CRYSTAL geometry
input keywords. For each keyword a brief explanation is reported as well
as whether the keyword requires a specific input.
Refer to CRYSTAL User's Manual for full
documentation.
| Symmetry information |
|
||
| SYMMOPS | printing of point symmetry operators | ||
| ATOMSYMM | printing of point symmetry at the atomic positions | ||
| Symmetry reduction |
|
||
| TRASREMO | removal of symmetry operators with translational components | ||
| SYMMREMO | removal of all symmetry operators | ||
| MODISYMM | removal of selected symmetry operators |
input
|
|
| Modifications without reduction of symmetry |
|
||
| ATOMORDE | reordering of atoms in molecular crystals | ||
| ORIGIN | shift of the origin to minimize the number of symmetry operators with translational components | ||
| PRIMITIV | crystallographic cell forced to be the primitive cell | ||
| REDEFINE | define a new cell with xy parallel to a given plane |
input
|
|
| Symmetry control |
|
||
| BREAKSYM | allow symmetry reduction following geometry modifications | ||
| KEEPSYMM | maintain symmetry following geometry modifications | ||
| Atoms and cell manipulation (possible symmetry reduction) |
|
||
| ATOMSUBS | substitution of atoms |
input
|
|
| ATOMREMO | removal of atoms |
input
|
|
| ATOMINSE | addition of atoms |
input
|
|
| ATOMDISP | displacement of atoms |
input
|
|
| ATOMROT | rotation of groups of atoms |
input
|
|
| SUPERCEL | generation of super cell |
input
|
|
| ELASTIC | distortion of the lattice |
input
|
|
| From crystals to slabs |
|
||
| SLABCUT | generation of a slab parallel to a given plane |
input
|
|
| From periodic structure to clusters |
|
||
| CLUSTER | cutting of a cluster from a periodic structure |
input
|
|
| Molecular crystals |
|
||
| MOLECULE | extraction of a set of molecules from a molecular crystal |
input
|
|
| MOLSPLIT | periodic structure of non interacting molecules | ||
| MOLEXP | variation of lattice parameters at constant symmetry and molecular geometry |
input
|
|
| BSSE correction |
|
||
| MOLEBSSE | counterpoise method for molecules (molecular crystals only) |
input
|
|
| ATOMBSSE | counterpoise method for atoms |
input
|
|
| Auxiliary and control keywords |
|
||
| ANGSTROM | Set input unit to Angstrom | ||
| BOHR | Set input unit to bohr | ||
| FRACTION | Set input unit to fractional | ||
| PARAMPRT | output of parameters controlling code dimensions | ||
| NEIGHBOR | number of neighbours in geometry analysis |
input
|
|
| ANGLES | bond angles and dihedral angles analysis |
input
|
|
| PRINTOUT | setting of printing options |
input
|
|
| RAYCOV | modification of covalent radii |
input
|
|
| SETPRINT | setting of printing options |
input
|
|
| TESTGEOM | stop after checking the geometry input | ||
| STOP | execution stops immediately | ||
| END | terminate processing of geometry input | ||
| Output of data on external units |
|
||
| COORPRT | output of the coordinates of all the atoms in the cell | ||
| EXTPRT | generation of input file for visualization | ||
| MOLDRAW | generation of input file for the program MOLDRAW | ||
| FINDSYM | generation of input file for the program findsym | ||
Note: the NEIGHBOR option requires
the full CRYSTAL input.
T. Hahn,
International Tables for Crystallography,
Reidel Publishing Company, 1987
T.C.W Mak, G.-D. Zhou,
Crystallography in Modern Chemistry
- A Resource Book of Crystal Structures,
Wiley, 1992
Research papers
R. Dovesi and R. Orlando
Convergence properties of the super cell approach in the study of local defects in solids
Phase Trans. 52, 151-167 (1994)
R. Orlando, R. Dovesi, P. Azavant, N.M.
Harrison and V.R. Saunders
A super-cell approach for the study of
localized defects in solids: carbon substitution in bulk silicon
J. Phys.: Cond. Matter 6, 8573-8583 (1994).
R. Orlando, P. Azavant, M.D. Towler, R.
Dovesi and C. Roetti
Cluster and super cell calculations for
carbon-doped silicon
J. Phys.: Cond. Matter 8, 1123-1133 (1996).
A. Lichanot, R. Orlando, G. Mallia, M.
Merawa, R. Dovesi
VOH center in magnesium
oxide: an ab initio super cell study
Chem. Phys. Lett. 318, 240 (2000)
R. Dovesi, R. Orlando, F. Ricca and C.
Roetti
CO adsorption on MgO crystals: Hartree-Fock
calculations for regular ad layers on a (001) lattice plane
Surface Sci. 186, 267-278 (1987).
R. Dovesi, M. Causa', R. Orlando, C. Roetti,
and V.R. Saunders
Ab initio approach to molecular crystals:
A periodic Hartree-Fock study of crystalline urea
J. Chem. Phys. 92, 7402 (1990).
C. Gatti, V.R. Saunders, C. Roetti
Crystal field effects on the topological
properties of the electronic density in molecular crystals: The case of
urea
J. Chem. Phys. 101, 10686 (1994)
|
|
|